
Zahra Mousavi 1; Elnaz Karbaschian 1; Ali Javadmanesh 1*
1, Department of Animal Science, Faculty of Agriculture, Ferdowsi University of Mashhad, Mashhad, Iran
E-mail:
javadmanesh@um.ac.ir
Received: 15/01/2024
Acceptance: 07/04/2024
Available Online: 08/04/2024
Published: 01/10/2024

Manuscript link
http://dx.doi.org/10.30493/DLS.2024.435863
Abstract
Nivalenol (NIV) is a prevalent mycotoxin that poses challenges for effective mitigation using conventional approaches, including the use of mineral and organic adsorbents. Hence, it is recommended to explore enzyme-based approaches for the detoxification of these specific mycotoxins, necessitating prior bioinformatics investigations to inform subsequent laboratory experiments. Thus, the objective of this research is to examine the affinity between DEP-A and DEP-B enzymes and NIV, as well as its oxidized form (3-keto-NIV), via molecular docking technique. The SWISS-MODEL server was utilized to predict the tertiary structure of the DEP-A and DEP-B proteins. Subsequently, an assessment was conducted on the stability of these structures under dynamic settings utilizing the GROMACS software. The H-DOCK web server was utilized to examine the interactions between the DEP-A enzyme and NIV, as well as between DEP-B and 3-keto-NIV. Subsequently, a molecular dynamics simulation was conducted in order to assess the stability of the linkages. Upon examination of the anticipated structures of the enzymes, it was observed that the predictions were accurate. The root mean square deviation (RMSD) curves indicate that both DEP-A and DEP-B achieved stability after 300 nanoseconds. The findings from the molecular docking analysis indicate that both DEP-A and DEP-B enzymes exhibited significant binding to their respective substrates, namely NIV and DEP-B with 3-keto-NIV. Furthermore, both enzyme-substrate complexes showed robust stability under dynamic settings. The findings suggest that both examined enzymes exhibited the capability to effectively attach to the NIV chemotype at the correct site, indicating their potential efficacy in deactivating this mycotoxin. Nevertheless, it is imperative to conduct laboratory investigations in order to validate these findings.
Keywords: Nivalenol, Mycotoxin, Devosia, Enzymes, Molecular docking
Introduction
Mycotoxins are a class of secondary metabolites synthesized by filamentous fungi. The toxic effects of mycotoxins are primarily associated with foodborne illnesses [1]. Trichothecenes, which are mycotoxins produced by many species of Fusarium fungi, are prevalent contaminants in animal feed across temperate zones in America, Europe, and Asia [2]. Fungal microorganisms release many forms of trichothecenes, namely type A (T-2 and HT-2 toxins) and type B (deoxynivalenol, nivalenol, and their derivatives). One of the most prevalent mycotoxins globally is Nivalenol (NIV), which has detrimental effects on both human health and the productivity of livestock [3]. The primary consequence of trichothecene mycotoxins is the early inhibition of cellular protein centers, followed by the cessation of DNA and RNA synthesis. Thus, the primary mechanism for toxicity mostly involves the inhibition of protein and macromolecule synthesis, as well as the suppression of the immune system. At lower concentrations, the presence of this mycotoxin induces symptoms such as nausea, diarrhea, ulcers, and lesions in the gastrointestinal tract, resulting in decreased food productivity and weight loss. Conversely, at higher concentrations, it elicits more severe effects including vomiting, significant weight loss, and impairments in the immune system [1].
Fusarium species are known to synthesize three significant groups of mycotoxins, specifically trichothecenes, fumonisins, and zearalenones, together with their associated mycoestrogens [4]. The production of mycotoxins by Fusarium fungi, including deoxynivalenol (DON) and nivalenol (NIV), has been documented by researchers [5]. Additional research has indicated that the toxicity issue associated with NIV is further exacerbated by its synergistic effects with DON, a member of the same mycotoxin family [6]. Consequently, the health hazards posed by both substances are heightened, necessitating a more intricate approach to their management.
A range of methodologies has been employed to address the issue of toxins, including the utilization of absorbent materials. Some of these materials showed potential in reducing the intake of mycotoxins, including NIV and DON [7]. However, the utilization of enzymes in handling mycotoxins issue appears more promising, since enzymes can modify the biological structure of mycotoxins, thereby converting them into molecules that are devoid of toxicity [8][9]. Enzymatic reactions frequently exhibit characteristics of irreversibility, efficacy, and environmental compatibility, hence avoiding the generation of any residual byproducts.
Currently, several microorganisms derived from soil, plants, and animal gastrointestinal tracts have been documented to possess the capacity to enzymatically modify the structural configuration of mycotoxins. The Nocardioides bacterium strain WSN05-2, which was isolated from soil samples collected from wheat fields, has the ability to alter the biological structure of DON and convert it into a non-toxic intermediate [10]. The research findings indicate that Devosia mutans 17-2-E-8 possess the capability to convert DON into the oxidized form (keto-DON-3), followed by its further transformation into the epimerized form (epi-DON-3). These forms are characterized by a spatial rearrangement of the OH-3 group [11]. The metabolites in question have demonstrated 10- and 50-times lower toxicity levels in both in vivo and in vitro settings [12][13]. The aforementioned works have identified ATP-binding protein [Devosia sp. 17-2-E-8] (DEP-A) and dehydratase [Devosia sp. 17-2-E-8] (DEP-B) as the enzymes responsible for the oxidation and epimerization of DON.
The application of genetic engineering techniques in the large-scale synthesis of enzymes, along with the utilization of bioinformatics expertise to forecast the interplay between enzymes and substrates, has emerged as a viable approach to mitigate expenses and enhance the efficiency, velocity, and precision of enzyme production. The utilization of computational techniques, such as molecular docking, to predict the efficacy of various compounds as potential therapeutic candidates prior to their production, expedites the drug discovery process and mitigates expenses [14][15]. Previous studies examined the molecular interaction of the mycotoxin DON on the crystal structure of DEP-A derived from Devosia mutans bacteria by in silico analysis [11][16]. However, the potential of DEP-A and DEP-B enzymes in NIV detoxification is yet to be investigated. The objective of this study is to utilize bioinformatic methods to predict the structure of DEP-A and DEP-B enzymes, and subsequently perform molecular docking experiments to assess the strength of the resulting complexes and binding sites between these enzymes and NIV, as well as its oxidized structure (3-keto-NIV), under in silico conditions.
Material and Methods
Preparation of DEP-A and DEP-B enzymes’ amino acid sequence
The amino acid sequences of DEP-A and DEP-B enzymes were obtained from the National Center for Biotechnology Information (NCBI) database under accession numbers KFL25551 and KFL28068. These sequences were utilized for protein tertiary structure prediction and assessment of physicochemical properties. The structure of NIV was acquired in SDF format from the PubChem database and subsequently converted to PDB format using the Open Babel software. ChemDraw software was employed to analyze the oxidized structure of NIV (3-keto-NIV).
DEP-A and DEP-B tertiary structure prediction
The SWISS-MODEL software was utilized to predict the tertiary structure of the DEP-A and DEP-B enzymes, while the Modeller software was employed to replace the missing amino acids. The accuracy and correctness of the constructed structures were assessed using SAVES v6.0 and ProSA servers, and the most appropriate structures were saved in PDB format.
Docking and molecular dynamics
Molecular dynamics (MD) simulation was employed to investigate the structures of DEP-A and DEP-B enzymes, utilizing appropriate structures selected from the preceding step. The simulation was conducted using the GROMACS software version 4/2/5, employing the modified amber 99sb force field and SPC216/E water molecules. The molecule of interest was positioned within the central region of the simulated cubic container, maintaining a separation of 1 nm from the container walls. Periodic boundary conditions were applied in the simulation. The system was neutralized by employing sodium and chlorine ions, taking into consideration the physiological conditions of the enzyme. The electrostatic energy of each periodic box was quantified using the Particle Mesh Ewald (PME) summation technique, while the estimation of non-bonded interactions (specifically electrostatic and van der Waals forces) was conducted using the Lennard-Jones model with a cut-off radius of 10°. The reduction of system energy was achieved through the implementation of the steepest descent algorithm, in conjunction with the utilization of the Berendsen coupling algorithm and the V-rescale thermostat. Prior to conducting the main simulation, the temperature (310 K) and pressure were coupled for a duration of 1 picosecond. Ultimately, the molecular dynamics (MD) simulation was executed in accordance with the aforementioned parameters.
To assess the stability of the predicted enzyme structure, root mean square deviation (RMSD) versus time plots were generated throughout the simulation duration (300 ns). The RMSD diagram is a metric that quantifies the positional deviation of one or more atoms, providing insight into the average inter-atomic distances within proteins. The stability of a protein structure is directly proportional to the magnitude of its deviations. A smaller RMSD value and a minimal fluctuation in this value indicates the stability of the protein model and an RMSD value below 2 Å is preferable in docking experiments as an indicator of the resulting complex stability. On the other hand, an increase in the RMSD or the slope of the graph indicates a higher degree of structural instability, leading to a faster disintegration of the structure [17].
The investigation of the interaction between the DEP-A enzyme and NIV, as well as DEP-B with 3-keto-NIV, was conducted using the H-DOCK online software. In this analysis, the enzyme was treated as the receptor, while the mycotoxin molecules were regarded as ligands. The assessment of appropriate structures was conducted by employing Pymol and PDBsum software to determine the lowest binding energy and proper orientation, thereby facilitating the observation of hydrogen bonds after molecular docking.
Results and Discussion
The tertiary structure of DEP-A and DEP-B enzymes
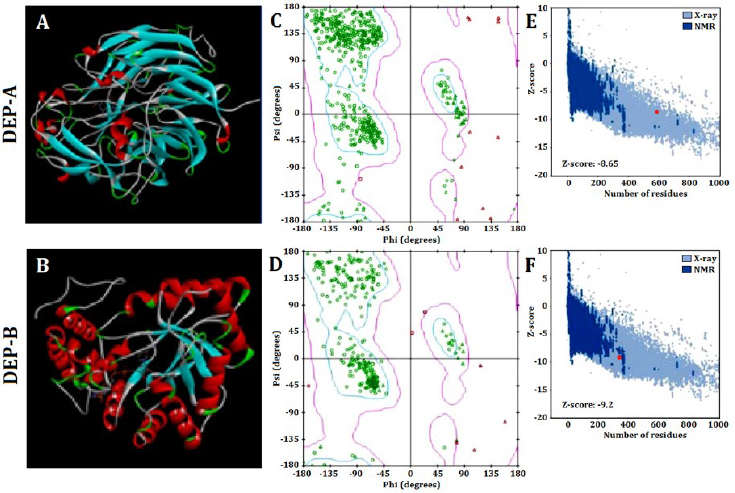
The SWISS-MODEL web server was utilized to predict the tertiary structure of DEP-A and DEP-B enzymes, which was subsequently modified using Modeller (Fig. 1 A and B). The evaluation of the prediction accuracy of the tertiary structure was conducted using multiple methodologies. Based on Ramachandaran’s analysis, it is observed that approximately 85% of the amino acids comprising the structures of DEP-A and DEP-B are situated within the intended region (Fig 1 C and D). This finding serves as an indication of the accuracy of the predicted tertiary structure. The Z-score values obtained from the ProSA server for DEP-A and DEP-B were -9.2 and -8.65, respectively. The NMR/X-ray plot of established structures demonstrated that the Z-score for DEP-A and DEP-B fell within the range of scores observed for native protein structures of comparable size (Fig. 1 E and F). The SAVES server obtained an ERRT evaluation of approximately 91% for DEP-A and 86% for DEP-B. ERRT is an internet-based server that validates the three-dimensional conformation of proteins by analyzing the nuclear connectivity among various atoms. A protein model that exhibits an ERRT value exceeding 80% is deemed to be a reliably predicted model, thereby validating the accuracy of the employed models for DEP-A and DEP-B enzymes.
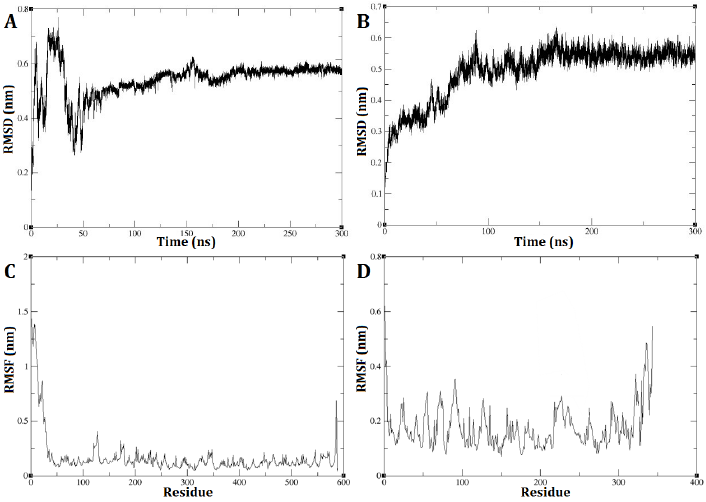
The GROMACS software was utilized to conduct a molecular dynamic analysis of the DEP-A and DEP-B enzymes. The RMSD diagram of the two enzymes, DEP-A and DEP-B, demonstrates that their respective structures achieved stability within a time frame of 300 nanoseconds. The RMSD is commonly employed as a metric to quantify the dissimilarity between the conformation of a protein in its initial state and its subsequent final position. This is due to the fact that the stability of a protein can be inferred from the proportion of deviations observed during its simulation.
DEP-A enzyme exhibited significant fluctuations in RMSD during the initial phase of the simulation, spanning the first 50 nanoseconds. Subsequently, reduced changes in RMSD were observed, ultimately reaching 0.58 nm between 200 and 240 nanoseconds (Fig. 2 A). As for DEP-B enzyme, the trajectory exhibited an initial upward trend during the simulation, which persisted until approximately 150 nanoseconds. Subsequently, from around 150 nanoseconds to 300 nanoseconds, the alterations in RMSD became more nuanced, ultimately converging at a value of approximately 0.55 nm (Fig. 2 B). This observation indicates the robustness and stability of these enzymes under the experimented conditions. The analysis of the root mean square fluctuation (RMSF) diagram revealed the probability of a correlation between the pronounced fluctuations of the RSMD and the absence of the N-terminal amino acids (specifically, the first 24 amino acids) of DEP-A that were missed in the initial model prediction with swiss model and then fixed with the Modeller (Fig. 2 C). On the other hand, the fluctuations in RMSF diagram of DEP-B were much more subtle (0.3 nm on average) (Fig. 2 D).
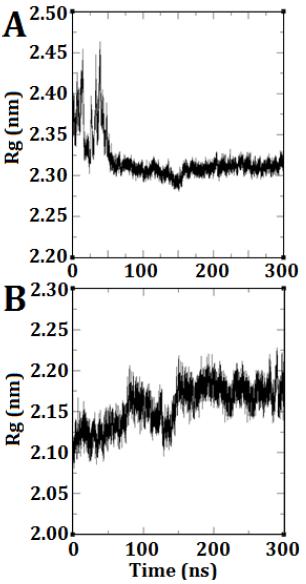
The radius of gyration refers to the average squared distance between the atoms within a molecule and its center of gravity. The plot provides a prediction of the degree of compression, expansion, and folding exhibited by a molecule throughout the simulation. In DEP-A, a notable fluctuation was observed at the onset of the simulation, resembling the RMSD diagram. However, this fluctuation gradually diminished over time, resulting in a consistent and horizontal trend (Fig. 3 A). This indicates that the anticipated structure achieved stability and did not undergo significant alterations. Regarding DEP-B, the structural expansion occurred gradually from its condensed state, exhibiting a gradual increase until it achieved stability at approximately 150 nanoseconds (Fig. 3 B).
Molecular docking
The present study utilized the H-DOCK server to conduct molecular docking. The calculation of connection scores is performed using a scoring function that relies on either ITScorePP or ITScorePR. The more negative value of the connection score, the greater the stability of the complex. An additional evaluative criterion present in this server is the confidence score. If the value falls within the range of 0.5 to 0.7, it suggests the potential for a connection between two molecules, whereas a score below 0.5 signifies a weak connection between the two molecules.
The confidence scores for the DEP-A+NIV and DEP-B+3-keto-NIV complexes were determined to be 0.57 and 0.65, respectively. The binding energies for the previously mentioned complexes were calculated to be -157.68 and -116.18, respectively. In the DEP-A+NIV complex, the hydrogen bond lengths at TYR439 and THR456 were measured to be 2.50 and 2.79 Å, respectively. Furthermore, it was observed that the amino acids Gln198, Asp311, Tyr454, Phe412, Leu413, Val453, and Leu441 of the PQQ (DEP-A cofactor) molecule exhibited hydrophobic interactions with NIV (Fig. 4 A). The interaction between the C3 of 3-keto-NIV and the DEP-B enzyme involved the formation of four hydrogen bonds with Tyr53, Lys81, His122, and Tyr241, with bond lengths measuring 3.19, 3.22, 2.77, and 2.19 Å, respectively. Within this intricate system, the molecules NADPH, Met21, Gln180, Trp206, and Arg244 predominantly engaged in hydrophobic interactions with 3-keto-NIV (Fig. 4 B).
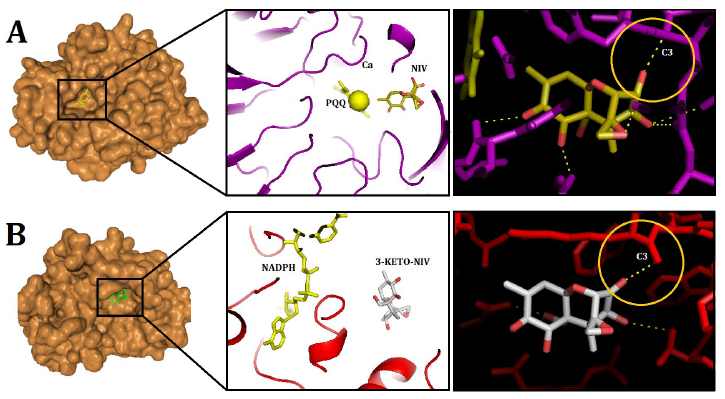
In a previous study, the molecular docking of deoxynivalenol (DON) with the crystal structure of Devosia albogilva DEP-A was conducted, yielding a binding energy of -6.56 kcal/mol [16]. According to the aforementioned study, it was observed that the hydroxyl group located at the C3 position, which is associated with the toxicity of DON, is in close proximity to PQQ and Ca2+, whereas the other two hydroxyl groups, which are not related, are relatively distant. This finding aligns with the fact that only the hydroxyl group at the C3 position undergoes conversion, thereby enhancing the credibility of the binding outcomes. These results are consistent with the findings obtained from the DEP-A+NIV docking analysis conducted in the present investigation. Moreover, the present study aligns with previous research by confirming that the hydrophobic bond in the DEP-A+DON complex consists of Leucine (Leu), Aspartic Acid (Asp), and Phenylalanine (Phe) amino acids [16]. It should be noted that dehydrogenase family enzymes possess an active site that comprises specific amino acids, including histidine (His), aspartic acid (Asp), threonine (Thr), and arginine (Arg). These amino acids are known to significantly contribute to the catalytic activity exhibited by these enzymes [18]. The aforementioned findings are consistent with the current observations regarding the participation of Asp and Thr in the connections associated with the DEP-A active site. The binding interface between PQQ and Ca2+ has been identified as Gln198, which aligns with previous research findings that identified Gln198 as the interface between DEP-A and 15A DON, a chemotype of DON [19]. Additionally, Gln171 has been proposed as the binding interface between PQQ and Ca2+.
NADH/NAD+ and NADPH/NADP+ function as a pair of redox cofactors within the cellular context. The involvement of the nicotinamide ring in these cofactors is direct in facilitating electron transfer during reactions catalyzed by NAD(P) H-dependent oxidoreductases. Additionally, the C4 carbon atom of the nicotinamide ring serves as a proton acceptor/donor [20]. According to a study on structural analysis, it was found that the residues Lys217, Arg290, and Gln294 play a crucial role in facilitating the binding of the DepBRleg enzyme (an enzyme analogous to DEP-B) to NADPH [21]. The catalytic residues Asp48, Tyr53, Lys81, and His122 exhibit conservation in the DepBRleg protein. Additionally, the substrate binding site encompasses the residues Met21, Asp48, Val52, Tyr53, Lys81, Arg83, Phe84, His122, Ala123, Ser152, Asn153, and Gln180. In a separate investigation, it has been documented that Oxid 15A DON forms a hydrophobic interaction with DEP-A through the involvement of specific amino acids, namely Met21 and Gln180. Additionally, Oxid 15A DON has been observed to establish a hydrogen bond with Lys81 and His122 [19]. In a similar vein, the findings of this study indicate that 3-keto-NIV formed hydrophobic interactions with the amino acids Met21 and Gln180, while establishing hydrogen bonds with Lys81, His122, and Tyr53. These observations suggest that the binding event occurred within the active site of the enzyme.
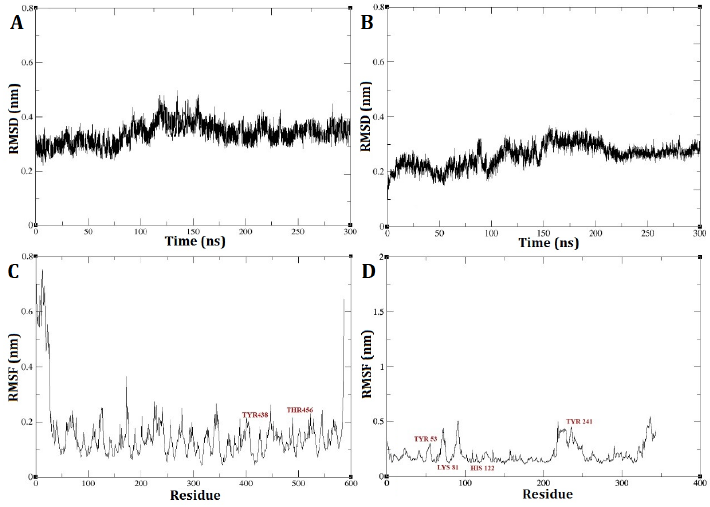
The RMSD outcomes for the DEP-A+NIV and DEP-B+3-keto-NIV complexes exhibited a state of relative stability throughout the entire 300 nanosecond simulation period (Fig. 5 A and B). A marginal, gradual alteration was observed in the simulation from the initial stage to the time intervals of 125 and 150 ns, pertaining to the DEP-A+NIV and DEP-B+3-keto-NIV complexes, respectively. This suggests the possibility of structural modifications occurring in the bond during this temporal span, such as bond formation or breakage. Nevertheless, the interaction between the complexes has achieved a state of stability, as evidenced by the RMSD of approximately 0.3 nm observed at the conclusion of the simulation. Typically, an increased quantity of robust hydrogen bonds leads to decreased RMSD variations, and a lower RMSD value indicates a higher level of stability in the intended system [22].
Upon analysis of the RMSF plot pertaining to the DEP-A+NIV complex, it was observed that there were discernible fluctuations of relative stability. Notably, the mobility was found to be elevated at the extremities of the C and N terminus of the enzyme. Examining the mobility of the regions associated with hydrogen bonds (TYR439 and THR456) revealed that these regions exhibited reduced mobility, suggesting a higher level of stability (Fig. 5 C). On the other hand, the RMSF diagram of the DEP-B+3-keto-NIV complex exhibited a marginally greater degree of fluctuation in comparison to both the DEP-A+NIV complex and DEP-B prior to docking. Nevertheless, upon conducting an individual investigation into the hydrogen bond regions located at Tyr53, Lys81, and His122 of the complex, it was observed that these regions exhibited limited mobility throughout the simulation. However, Tyr241 displayed a relatively higher RMSF value (Fig. 5 D). Hence, based on the consistent RMSD values observed during the simulation and the minimal RMSF values observed in the hydrogen bond regions, it can be concluded that both complexes exhibited a relatively stable behavior. Furthermore, it can be inferred that DEP-A and DEP-B effectively formed binding interactions with NIV and 3-keto-NIV, respectively.
Conclusion
The findings of the current investigation demonstrate the successful determination of the structural characteristics of DEP-A and DEP-B. The findings from the molecular docking analysis indicate that both DEP-A and DEP-B enzymes exhibit a high affinity for the substrate, as evidenced by their strong binding energies. The current research suggests that these enzymes may exhibit efficacy against both NIV and its oxidized structure (3-keto-NIV). However, it is important to note that further laboratory investigations are necessary to validate these findings.
References
| 1 | Liu M, Zhao L, Gong G, Zhang L, Shi L, Dai J, Han Y, Wu Y, Khalil MM, Sun L. Invited review: Remediation strategies for mycotoxin control in feed. J. Anim. Sci. Biotechnol. 2022;13(1):1-6. DOI |
| 2 | Creppy EE. Update of survey, regulation and toxic effects of mycotoxins in Europe. Toxicol. Lett. 2002;127(1-3):19-28. DOI |
| 3 | Kumar P, Mahato DK, Gupta A, Pandey S, Paul V, Saurabh V, Pandey AK, Selvakumar R, Barua S, Kapri M, Kumar M. Nivalenol Mycotoxin Concerns in Foods: An Overview on Occurrence, Impact on Human and Animal Health and Its Detection and Management Strategies. Toxins. 2022;14(8):527. DOI |
| 4 | Ekwomadu TI, Akinola SA, Mwanza M. Fusarium mycotoxins, their metabolites (free, emerging, and masked), food safety concerns, and health impacts. Int. J. Environ. Res. Public Health. 2021;18(22):11741. DOI |
| 5 | Yan P, Liu Z, Liu S, Yao L, Liu Y, Wu Y, Gong Z. Natural occurrence of deoxynivalenol and its acetylated derivatives in Chinese maize and wheat collected in 2017. Toxins. 2020;12(3):200. DOI |
| 6 | Cheat S, Pinton P, Cossalter AM, Cognie J, Vilariño M, Callu P, Raymond-Letron I, Oswald IP, Kolf-Clauw M. The mycotoxins deoxynivalenol and nivalenol show in vivo synergism on jejunum enterocytes apoptosis. Food Chem. Toxicol. 2016;87:45-54. DOI |
| 7 | Avantaggiato G, Havenaar R, Visconti A. Evaluation of the intestinal absorption of deoxynivalenol and nivalenol by an in vitro gastrointestinal model, and the binding efficacy of activated carbon and other adsorbent materials. Food Chem. Toxicol. 2004;42(5):817-24. DOI |
| 8 | Fang QA, Du M, Chen J, Liu T, Zheng Y, Liao Z, Zhong Q, Wang L, Fang X, Wang J. Degradation and detoxification of aflatoxin B1 by tea-derived Aspergillus niger RAF106. Toxins. 2020;12(12):777. DOI |
| 9 | Qiu T, Wang H, Yang Y, Yu J, Ji J, Sun J, Zhang S, Sun X. Exploration of biodegradation mechanism by AFB1-degrading strain Aspergillus niger FS10 and its metabolic feedback. Food Control. 2021;121:107609. DOI |
| 10 | Ikunaga Y, Sato I, Grond S, Numaziri N, Yoshida S, Yamaya H, Hiradate S, Hasegawa M, Toshima H, Koitabashi M, Ito M. Nocardioides sp. strain WSN05-2, isolated from a wheat field, degrades deoxynivalenol, producing the novel intermediate 3-epi-deoxynivalenol. Appl. Microbiol. Biotechnol. 2011;89:419-27. DOI |
| 11 | He JW, Bondy GS, Zhou T, Caldwell D, Boland GJ, Scott PM. Toxicology of 3-epi-deoxynivalenol, a deoxynivalenol-transformation product by Devosia mutans 17-2-E-8. Food Chem. Toxicol. 2015;84:250-9. DOI |
| 12 | Carere J, Hassan YI, Lepp D, Zhou T. The enzymatic detoxification of the mycotoxin deoxynivalenol: Identification of DepA from the DON epimerization pathway. Microb. Biotechnol. 2018;11(6):1106-11. DOI |
| 13 | Carere J, Hassan YI, Lepp D, Zhou T. The identification of DepB: An enzyme responsible for the final detoxification step in the deoxynivalenol epimerization pathway in Devosia mutans 17-2-E-8. Front. Microbiol. 2018;9:1573. DOI |
| 14 | Javadmanesh A, Mohammadi E, Mousavi Z, Azghandi M, Tanhaiean A. Antibacterial effects assessment on some livestock pathogens, thermal stability and proposing a probable reason for different levels of activity of thanatin. Sci. Rep. 2021;11(1):10890. DOI |
| 15 | Mousavi Z, Rashidian Z, Zeraatpisheh Y, Javadmanesh A. Molecular docking of bacteriocin enterocin P peptide with mastitis-causing E. coli antigen in cattle. Vet. Res. Biol. Prod. 2022;35(4):114-22. DOI |
| 16 | Yang H, Yan R, Li Y, Lu Z, Bie X, Zhao H, Lu F, Chen M. Structure–Function Analysis of a Quinone-Dependent Dehydrogenase Capable of Deoxynivalenol Detoxification. J. Agric. Food Chem. 2022;70(22):6764-74. DOI |
| 17 | Mousavi SZ, Javadmanesh A. Molecular docking of Enterocin-P peptide with DNA: an in silico study. In The 1st International and the 10th National Iranian Conference on Bioinformatics. 2022. |
| 18 | Farhana A, Lappin SL. Biochemistry, lactate dehydrogenase. StatPearls. 2022. |
| 19 | Mousavi Z, Sekhavati MH, Farzaneh M, Javadmanesh A. Investigating of the binding energy of DEP-A and DEP-B enzymes with DON mycotoxin chemotype by molecular docking. Vet. Res. Biol. Prod. 2023. DOI |
| 20 | Vidal LS, Kelly CL, Mordaka PM, Heap JT. Review of NAD (P) H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta Proteins Proteom. 2018;1866(2):327-47. DOI |
| 21 | Abraham N, Schroeter KL, Zhu Y, Chan J, Evans N, Kimber MS, Carere J, Zhou T, Seah SY. Structure–function characterization of an aldo–keto reductase involved in detoxification of the mycotoxin, deoxynivalenol. Sci. Rep. 2022;12(1):14737. DOI |
| 22 | Kony DB, Hünenberger PH, Van Gunsteren WF. Molecular dynamics simulations of the native and partially folded states of ubiquitin: Influence of methanol cosolvent, pH, and temperature on the protein structure and dynamics. Protein Sci. 2007;16(6):1101-18. DOI |
Cite this article:
Mousavi, Z., Karbaschian, E., Javadmanesh, A. The interaction between DEP-A and DEP-B enzymes with nivalenol mycotoxin and its oxidized structure: A bioinformatic investigation. DYSONA – Life Science, 2024;5(2): 52-61. doi: 10.30493/dls.2024.435863